

In the last few years, we have seen the introduction of increasing numbers of drugs manufactured as biological molecules and as Biosimilars. These drugs present special challenges both in their development and also in the approach to the target markets and their reg istration processes. Many of the biosimilar molecules come from India where approximately 50% of the global biosimilar companies are based and whe re a similar proportion of the world’s biosimilars are produced.
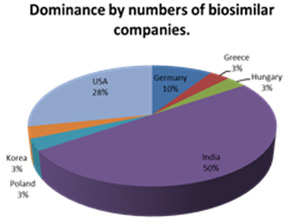
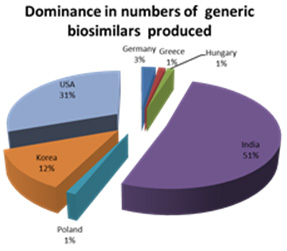
In terms of numbers of molecules, third rated after India and the United States is South Korea which produces approximately 12% of the biosimilar species whilst only having 3% of the Worlds Biosimilar companies.
The implication is that Asia is globally the geography where most of the world’s biosimilars and biological molecules will be produced in the near future and in terms of manufacturing and drug development, it is the regulations of these countries which will apply whe reas the market place for the biosimilars and biologicals is global. Hence any organisation such as ours, needs to be able to both work withi n Asia but be equally able to relate to the various needs of the target countries in terms of preparing the marketing dossier and the subsequent af ter-care once the molecule has been accepted for distribution within a particular country. Whereas for small molecules, there are close simil arities and concordance between the countries as to the requirements for registration, the new character of the biosimilars and biologic al means that considerable insight and knowledge is needed to make sure that the development and pharmacovigilance needs are met in much mo re diverse regulatory environments. It is repeatedly stated that biosimilars can never be considered as generics but each biosimilar has to be evaluated in a process more akin to a New Chemical Entity because even small differences in structure and manufacture may have large impli cations for safety and clinical activity. This was spelt out in s recent report when a pharmaceutical contributor (Genentech, owned by R oche) has taken the position that. “the properties of the biologic often depend directly on the nature of the manufacturing process. Furthermore, proteins have unique structural organization patterns (referred to as “folding”) that affect the way that they work in the body; even biolo gics that are chemically the same may have differing biological effects due to differences in the structural folding.
An example of this folding effect is the difference between a raw egg and a cooked one: chemically the two are the same, but they are physically and biologically very different.”11 The company supports clinical trials for each biosimilar indication. In the same review. Amgen who registered the first biosimilar in the EU, in testimony before the FDA, asserted that half of the biosimilars developed in Europe had unexpected clinical outcomes, and therefore relying on pharmacokinetic and pharmacodynamic studies al one should never be enough.
From an economic standpoint, the critical issue in biologics, as opposed to small molecules, may be in convincing doctors, pa tients and other healthcare stakeholders that the follow-on has demonstrated sufficient similarity to risk adopting it over the pioneer product.
Structuring a development programme for maximum marketing effectiveness. Companies producing these molecules may seek optimisation of their development and regulatory programmes so as to effectively market their pharmaceuticals to as many countries as possible with a single package which is designed from the beginning to meet as many d ifferent regulatory authorities as possible in a single acceptable programme. Typically a biosimilar biologics manufacturing company would wish to market their pharmaceutical in the USA, EU and then as many other jurisdictions as possible. This is further complicated by the desire of generic manufacturers to produce not only Biosimilars but that category of Biosimilar which is regarded as ‘an interchangable’. An i nterchangeable product must meet all the same requirements as a biosimilar and in addition have the same route of administration, dosage for m and strength as the reference product. An interchangeable “may be substituted for the reference product without the intervention of the heal th care provider who prescribed the reference product.”
In 2010, the United States enacted the Biologics Price Competition and Innovation Act (BPCI) which defined the path for the r egistration of Biosimilars. The European pathway had existed since 2005 and the arrival of the US brought both jurisdictions very close tog ether but with a number of significant differences. Within the US act, the biosimilar is well defined as:
(A) Highly similar to the reference product notwithstanding minor differences in clinically inactive components; and
(B). No clinically meaningful differences between the biological product and the reference product in terms of the safety, pu rity, and potency of the product.”
Protection to the original manufacturer of the biological is afforded for 12 years during which time the FDA will not grant a biosimilar application until the expiry of the of the original biologic license.